
Hemoglobinopathieën
Betrokken genen: HBA1, HBA2, HBB
Korte omschrijving: Sikkelcelziekte en alfa- en bèta- thalassemie zijn de belangrijkste, autosomaal recessief overervende, vormen van ernstige erfelijke bloedarmoede, ook wel hemoglobinopathieën (HbP-en) genaamd.
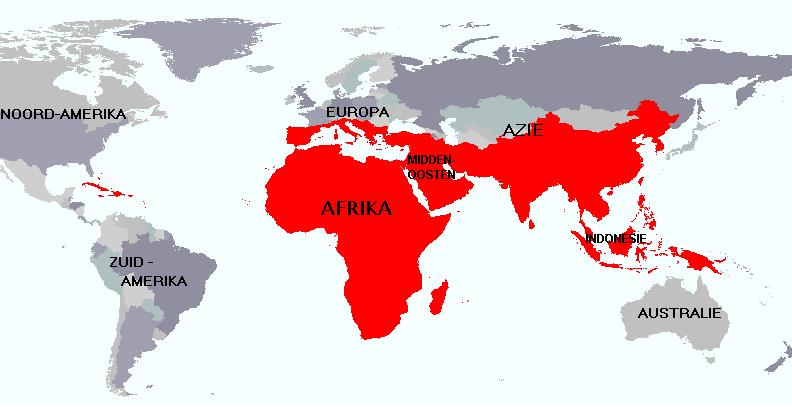
De kans op dragerschap is sterk verhoogd als (dragerschap) van sikkelcelziekte of thalassemie in de familie voorkomt, maar ook als (voorouders van) uw patiënt(en) oorspronkelijk afkomstig zijn uit Afrika, de Antillen, het Middellandse Zeegebied, Zuidoost-Azië of het Midden-Oosten (zie kaart in rood aangegeven). In deze gebieden varieert de dragerschapsfrequentie tussen 1 op 15 tot 1 op 7. In sommige Afrikaanse landen is zelfs 30-40% van de bevolking drager.
Let op: veel Surinamers en Antillianen hebben voorouders uit Afrika of Azië; sommige van hen hebben juist voorouders die oorspronkelijk uit Europa komen.
Zowel sikkelcelziekte als thalassemie kunnen ernstige bloedarmoede veroorzaken vanaf de zuigelingenleeftijd. Bij beide aandoeningen is behandeling in een zo vroeg mogelijk stadium van groot belang om vroege sterfte en morbiditeit zoveel mogelijk te vermijden.
Twee ‘gezonde’ dragers vormen samen een dragerpaar. Voor een dragerpaar bestaat er bij iedere zwangerschap een kans van 25% om een kind met een ernstige vorm van bloedarmoede te krijgen. Hierbij is het belangrijk te beseffen dat niet alleen als beide ouders drager zijn van sikkelcelziekte er een sterk verhoogde kans bestaat van 25% op het krijgen van een kind met sikkelcelziekte, maar dat dit ook kan gebeuren als één ouder drager is van sikkelcelziekte en de andere ouder van bèta-thalassemie.
-
Meer informatie
- OMIM
- GeneReviews
- Orphanet
- Informatiebrief
- Aanbevelingen en interpretatie van HbP onderzoek van de Vereniging Hematologisch Laboratoriumonderzoek
- Website van het Hemoglobinopathieën Laboratorium van het LUMC
- Richtlijn Preconceptie Dragerschapsonderzoek (PDO) voor hoogrisicogroepenParen of individuen afkomstig uit endemische malaria gebieden (Afrika, Suriname, de Antillen, landen rondom Middellandse Zee (bijvoorbeeld Turkije of Marokko), Zuidoost-Azië of het Midden-Oosten) hebben een verhoogd risico op dragerschap van hemoglobinopathieën.
-
Voor patiënten
- Erfelijkheid.nl
- Informatiebrief
- MedlinePlus Genetics: