
Verschillende genetische testen - Uitgebreid
Hieronder treft u een uitgebreide beschrijving van de verschillende technieken die in de genetische diagnostiek worden gebruikt. Er is ook een pagina met een beknopte beschrijving van de verschillende technieken en wat hiermee wel en niet kan worden aangetoond.
-
Onderzoek van chromosomen met array diagnostiek
Met de zogeheten micro-array analyse kan met steeds toenemende resolutie het gehele genoom in kaart worden gebracht.
Bij de array CGH (CGH = comparative genomic hybridization) wordt het enkelstrengs DNA van een patiënt samen met enkelstrengs controle DNA toegevoegd aan een microarray. Een microarray is een oppervlak (bijvoorbeeld een glaasje of een druppel vloeistof) met daaraan op vaste plaatsen verbonden bekende stukjes van enkelstrengs DNA. Het controle DNA en DNA van de patiënt beconcurreren elkaar voor binding aan dit DNA. Door het DNA van de patiënt en het controle DNA hun eigen fluorescente label te geven, is het mogelijk om, door het vaststellen van de verhouding tussen de fluorescente signalen, de verhouding tussen het aantal kopieën van het DNA van de patiënt en controle DNA vast te stellen. Zo kan worden onderzocht of er grote of kleine stukken van chromosomen ontbreken (deletie) of teveel aanwezig zijn (duplicatie). Deleties en duplicaties van 20 – 50 kb en groter kunnen hiermee tegenwoordig worden opgespoord. De array CGH is in de hedendaagse praktijk grotendeels vervangen door de SNP array.
Een andere array methode is de zogeheten SNP array. Een SNP (single nucleotide polymorfisme) is een (veelal onschuldige) variatie in het genoom van één enkele nucleotide. Het zijn plaatsen in het DNA die tussen individuen kunnen verschillen. Voor een gegeven SNP zijn er 2 mogelijke opties voor het aanwezige nucleotide (theoretisch 4, want er zijn immers 4 verschillende nucleotiden, maar in de praktijk meestal 2). Het meest voorkomende nucleotide noemt men allel A, het alternatieve nucleotide allel B. Omdat een individu 2 kopieën van elk allel heeft, kan je voor een gegeven nucleotide 2 keer allel A hebben (AA), allel A en B (AB) of 2 keer allel B. Met een SNP-array wordt bepaald welk allel van een gegeven SNP aanwezig is. Een SNP-array bepaalt dit voor vele tien- tot honderdduizenden SNPs verspreid over het genoom tegelijk.
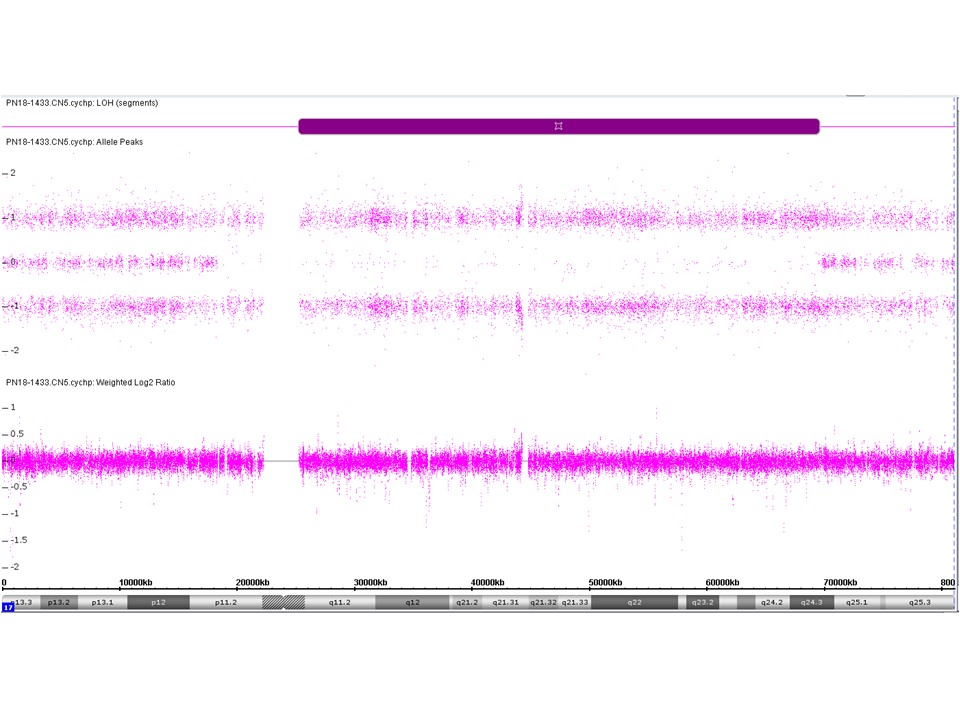
Voorbeeld van een SNP array resultaat van chromosoom 17. In het bovenste deel staat op de Y-as de B allele frequency (BAF). Een BAF van 0 correspondeert met een AB genotype, een BAF van 1 met BB en een BAF van -1 met AA. Normaal gesproken verwacht je bij iemand een willekeurige verdeling van AA, AB en BB. Als op een groot stuk van het chromosoom alleen maar 1 type SNP wordt gevonden (alleen AA of BB) duidt dit op afwezigheid van heterozygotie (absence of heterozygosity = AOH). Dat kan het geval zijn bij een deletie (je meet maar 1 type allel, omdat het andere allel niet aanwezig is) of een situatie waarbij een normaal kopie aantal van 2 is vastgesteld, maar dat de SNP’s van het betreffende chromosoom afkomstig zijn van 1 en dezelfde ouder. Er kan dan sprake zijn van uniparentale disomie. Indien bij meerdere chromosomen grote, homozygote stukken (Regions Of Homozygosoty = ROH) worden gedetecteerd, is er sprake van bloedverwantschap tussen de ouders van de onderzochte persoon. Het onderste deel geeft op de Y-as de Log2 R ratio. Dit geeft de verhouding tussen het aantal kopieën van de patiënt versus controle weer. In dit geval zijn die verhoudingen gelijk en zijn er geen aanwijzingen voor de aanwezigheid van een deletie: het betreft een normaal kopie aantal van 2 voor chromosoom 17. Een SNP array geeft per meetpunt, net als een array-CGH, informatie over het aantal kopieën van het gegeven stukje DNA dat bij een patiënt aanwezig is en of er van een gegeven stukje van beide ouders 1 kopie is.
Arrays zijn verrijkt voor coderende sequenties, zodat de resolutie voor genen hoog is. Array onderzoek is vooralsnog echter niet (altijd) geschikt om nucleotide varianten (veranderingen in 1 of enkele baseparen) op te sporen. Daartoe is Sanger sequencing of WES aangewezen.
-
Sanger sequencing (onderzoek van 1 of enkele genen)
Bij Sanger sequencen (sequencen = het bepalen van de basepaar/nucleotide volgorde van een stuk DNA) gebruikt men het enkelstrengs DNA van een patiënt als mal. Deze mal wordt met een DNA primer, DNA polymerase en nucleotiden (de bouwstenen van het DNA, Adenosine (A), Guanine (G), Cytosine (C) en Thymine (T)) verdeeld over 4 verschillende reacties. Aan elk van deze 4 reacties is 1 type gemodificeerde nucleotide toegevoegd (dus of A, of C, of G, of T). Deze nucleotiden hebben, naast dat ze gelabeld (meestal fluorescent) zijn, de eigenschap dat als ze worden ingebouwd er geen volgende nucleotide kan worden aangeplakt. Er worden dus telkens kopieën van het stukje DNA van de patiënt gemaakt die op een willekeurige plek worden afgebroken. M.b.v. elektroforese worden die stukjes op lengte gesorteerd en kan de volgorde van het DNA worden bepaald (zie afbeelding).
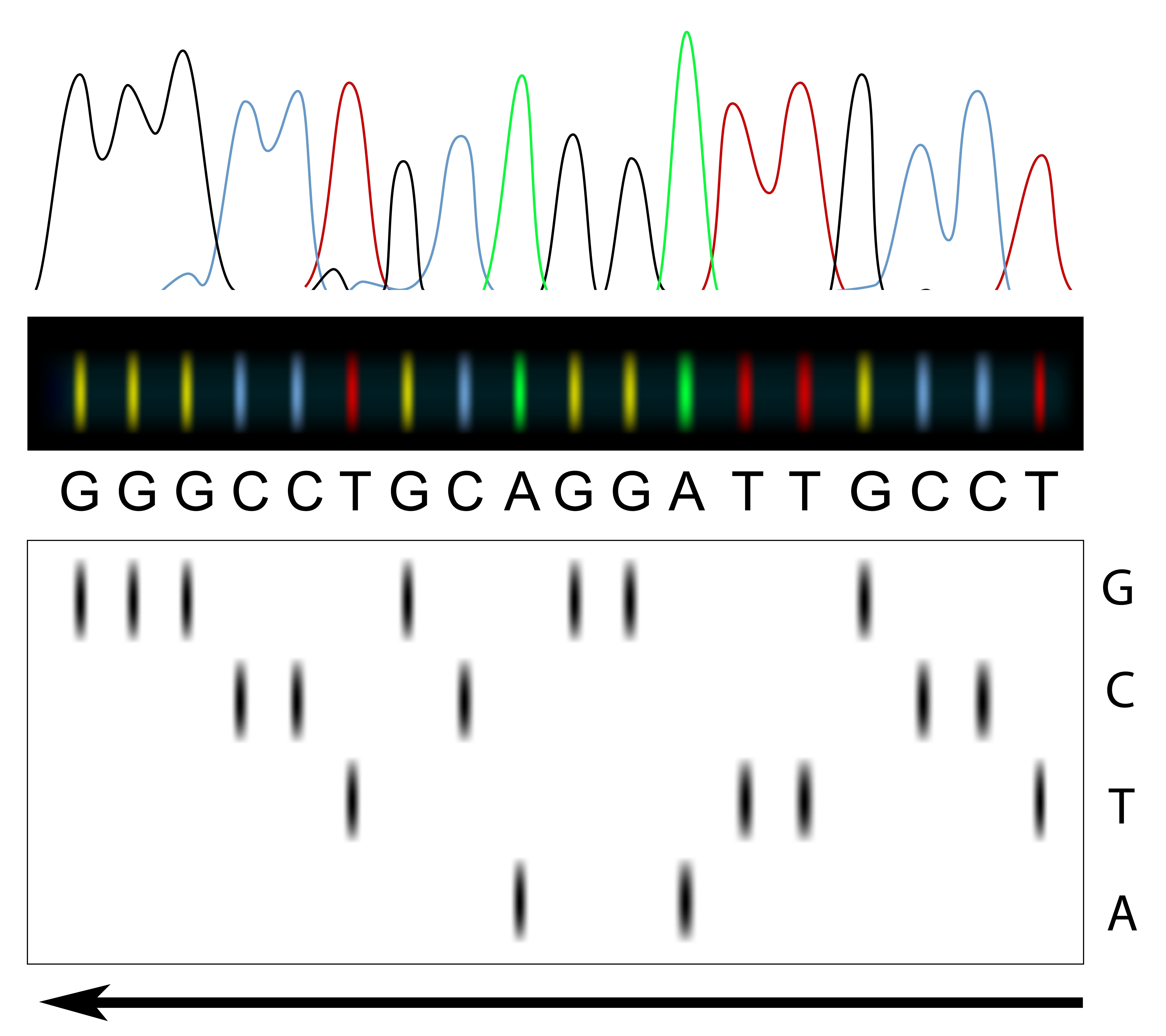
Hiermee kunnen DNA stukken van 500 tot 1000 bp worden afgelezen. Per keer kunnen er 96 reacties parallel worden verricht waarmee zo’n 115.000 bp per dag kunnen worden bepaald. Met sequentie analyse krijg je informatie over eventuele veranderingen in de aanwezige baseparen, maar deze analyse is niet geschikt om deleties en/of duplicaties mee aan te tonen.
-
Next generation sequencing
Next generation sequencing (NGS) is een verzamelnaam voor verschillende platformen en methoden van sequencen die gemeenschappelijk hebben dat er, i.t.t. Sanger sequencing, meerdere DNA fragmenten tegelijkertijd worden gesequenced. Met NGS kan op die manier van vele stukken DNA, parallel de volgorde worden bepaald.
Het DNA van een patiënt wordt hiertoe in kleine stukken opgeknipt en die kleine fragmenten worden met een adapter aan een oppervlak bevestigd. Zo’n adapter dient als primer (=startpunt) voor de sequencing reactie. Afhankelijk van de NGS techniek worden de DNA fragmenten direct gesequenced of eerst vermenigvuldigd (waarmee locaal kloons van het fragment ontstaan) en dan gesequenced. Het sequencen kan vanaf 1 uiteinde van het DNA fragment plaatsvinden (single-end read) of van beide uiteinden (paired-end read).
Sequentiedata worden per nucleotide dat wordt toegevoegd aan elk DNA fragment gegenereerd i.p.v. na elektroforese. Dit proces verloopt automatisch en sneller dan bij Sanger sequencing. De lengte van de fragmenten (=read length) die op die manier kan worden gesequenced is momenteel gemiddeld zo’n 30-400 bp (van een DNA fragment van bijvoorbeeld 1000 bp zal met deze methode alleen de sequentie van de eerste 30-400 bp worden bepaald). Dat zijn dus kortere stukken dan per keer met Sanger sequencing worden afgelezen, maar met NGS worden er veel van deze kleine fragmenten tegelijkertijd gesequenced.
De sequencing data van al die kleine fragmenten wordt gecombineerd en gekoppeld aan de juiste positie op het normale referentie genoom. Bij een korte read length kan een fragment in theorie passen op meerdere posities op het referentiegenoom.
Een willekeurig basepaar wordt met NGS vele malen gesequenced (sequentie diepte/sequencing depth/verticale dekkingsgraad). Dat is ook nodig, omdat er bij het sequencen fouten gemaakt kunnen worden die je wilt kunnen onderscheiden van een ware verandering in de sequentie. Om betrouwbaar te kunnen vaststellen welk basepaar op een bepaalde plek in het genoom aanwezig is, wil je dus meerdere keren bepaald hebben welk basepaar daar aanwezig is. Zo onderscheidt je een sequencing fout van een ware verandering in de DNA sequentie.
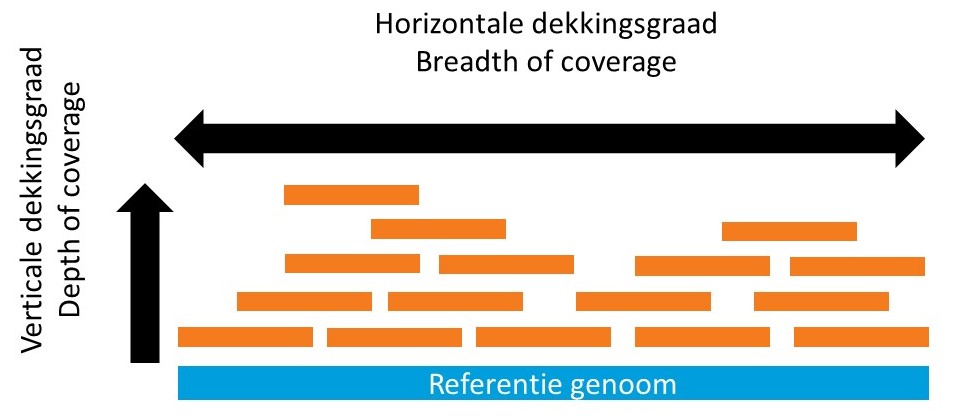
De gemiddelde dekking (of coverage) van een NGS experiment geeft aan hoe vaak iedere nucleotide van het afgelezen stuk DNA is gesequenced. De dekkingsgraad kan echter verschillen per gen. Het kan zo zijn dat bepaalde delen van het genoom heel vaak worden gesequenced, terwijl andere delen niet of nauwelijks worden gesequenced. Met name gebieden die veel repeats (zich herhalende korte DNA fragmenten) bevatten, worden vaak minder goed afgelezen. De breedte (= breadth = horizontale dekkingsgraad) van een NGS experiment geeft aan welk percentage van het geanalyseerde stuk DNA tenminste een gegeven aantal keer is gesequenced.
NGS kan ook worden gebruikt om informatie te krijgen over aanwezigheid van deleties of duplicaties. Wanneer er sprake is van een deletie of duplicatie dan is de sequentie diepte voor dat stuk van het genoom lager respectievelijk hoger. Dit vereist echter een bepaalde technische infrastructuur en substantiële bio-informatische ondersteuning, die op dit moment nog niet overal op routinebasis gerealiseerd zijn.
Paired-end sequencing kan gebruikt worden om inserties en deleties nauwkeuriger vast te stellen. Voor het NGS experiment selecteer je DNA fragmenten waarvan je de lengte weet. Daarmee weet je dus ook de afstand tussen de 2 uiteinden van het DNA fragment. Door vervolgens de afstand te vergelijken met de afstand na mappen met het referentiegenoom krijg je informatie over eventueel aanwezige deleties of duplicaties.
Vooralsnog is array onderzoek echter betrouwbaarder om genoomwijd deleties en duplicaties op te sporen.
-
WES / WGS (onderzoek van alle genen of een genpakket(=WES met filter))
De NGS techniek kan worden toegepast voor WES onderzoek. WES staat voor whole exome sequencing. Hiermee wordt de sequentie van ~95% van de eiwitcoderende sequenties in 1 experiment bepaald. Dit levert een grote hoeveelheid data op die in stappen wordt geanalyseerd. Bij targeted WES (panel/pakketanalyse) worden alleen de genen waarvan bekend is dat zij geassocieerd zijn met een gegeven soort aandoening geanalyseerd. Bij een open WES ofwel exoomwijde aanvraag worden alle genen geanalyseerd.
WES onderzoek kan veel varianten in genen opleveren. Bij een WES analyse i.v.m. ontwikkelingsachterstand bij een kind wordt daarom vaak ook het DNA van de ouders geanalyseerd (trio analyse). Op die manier kan aan veel varianten gelijk betekenis worden gegeven: is een variant bij één van beide (gezonde) ouders aanwezig, dan is het minder waarschijnlijk dat dit de verklaring is voor een ontwikkelingsachterstand bij het kind. Is een variant nieuw (‘de novo’) ontstaan bij het kind, dan is dit in het algemeen meer waarschijnlijk. Met deze trio analyse kunnen echter ook pathogene varianten opgespoord worden die bij een recessieve of X-gebonden overervingsvorm van de aandoening bij de patiënt zouden kunnen passen. WES onderzoek kan varianten met een nog onzekere/ onbekende betekenis voor het ziektebeeld opleveren (Variant of Unknown Significance).
Bij een open WES kunnen daarnaast pathogene varianten worden geïdentificeerd die niet samen hangen met de oorspronkelijke vraag, maar wel geassocieerd zijn met een andere aandoening (nevenbevinding). Het is belangrijk de kans op nevenbevindingen en het beleid hier omtrent van tevoren te bespreken. Open WES kan om die reden vooralsnog alleen door een klinisch geneticus worden aangevraagd, omdat de kans op nevenbevindingen relatief groot is.
Bij whole genome sequencing (WGS) worden niet alleen alle eiwitcoderende sequenties van het DNA onderzocht, maar ook de stukken van het DNA die niet voor eiwit coderen.
-
Klassiek chromosomenonderzoek: karyogram en FISH (fluorescente in situ hybridisatie)
Het klassieke chromosomenonderzoek bestaat uit het vervaardigen van een afbeelding van de chromosomen zoals deze tijdens de metafase van de celdeling te zien zijn onder de microscoop (een karyogram). Hierbij worden de chromosomen gerangschikt naar grootte. Om een karyogram te kunnen maken, worden cellen aangezet tot delen; zodra cellen de metafase bereiken, worden ze stilgelegd en chemisch gekleurd. Door de kleuring ontstaat een goed zichtbaar bandenpatroon op de chromosomen. Door een karyogram te bestuderen kunnen afwijkingen in aantal of vorm van chromosomen vastgesteld worden. Alleen grotere afwijkingen (5- 10 Mb en groter) kunnen hiermee worden opgespoord.
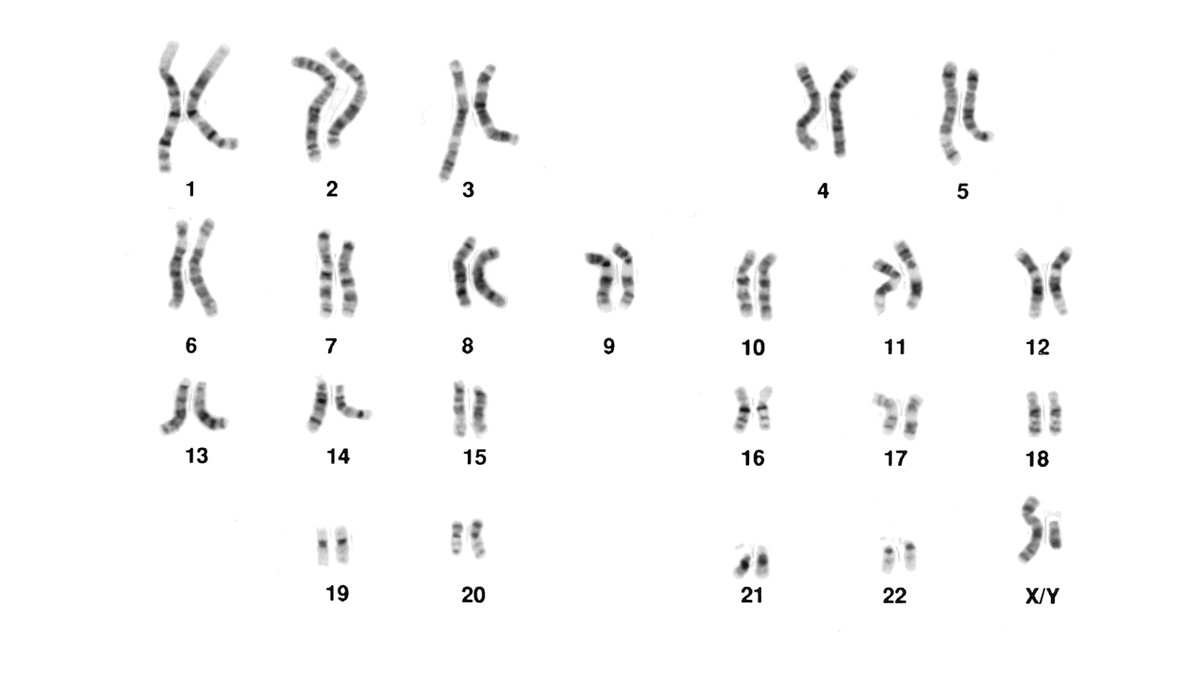
Klassiek microscopisch chromosomenonderzoek in de vorm van een karyogram is alleen nog aangewezen bij sterke verdenking op een numerieke afwijking zoals Down syndroom of Turner syndroom. Met een karyogram kan men nagaan waar de numerieke afwijking zich bevindt (en dus differentiëren tussen een vrije trisomie en een translocatie). Met array onderzoek is dit niet mogelijk.
FISH (fluorescente in situ hybridisatie) wordt gebruikt in aanvulling op het karyogram. Bij FISH onderzoek wordt een bekend stuk DNA dat complementair is aan een bepaald stuk DNA waarin men geïnteresseerd is fluorescent gelabeld. Dit wordt toegevoegd aan het DNA van de patiënt. Vervolgens kan onderzocht worden of de fluorescente probe bindt, waar en hoe vaak. FISH onderzoek wordt nog toegepast om bijvoorbeeld na het vinden van een ogenschijnlijke de novo deletie of duplicatie bij een kind een complexe gebalanceerde afwijking bij één van de ouders uit te sluiten. Van belang is dat voor chromosomenonderzoek en / of FISH, delende cellen nodig zijn en dat om deze reden het bloed moet worden opgevangen in een Lithium/heparine buis (groene dop).
-
Onderzoek van imprintingsdefecten / afwijkende hypermethylatie
Methylering is het proces waarbij een methylgroep aan een DNA molecuul wordt gekoppeld. Dit leidt tot structuurveranderingen van het DNA waardoor de transcriptie van genen wordt beïnvloedt (en genen dus “aan” of “uit” worden gezet). Er zijn vele verschillende soorten aandoeningen die het gevolg zijn van afwijkingen in de methylering.
Imprinting is een proces waarbij het tot expressie komen van een gen afhankelijk is van de ouder van wie het gen geërfd is. Dit proces zorgt ervoor dat van een deel van de genen selectief alleen het maternale óf juist alleen het paternale allel tot uitdrukking komt. Deze imprinting blijft gedurende het hele leven aanwezig. Bij een klein aantal ziektebeelden kunnen afwijkingen in dit proces een rol spelen (bijvoorbeeld bij Angelman syndroom/Prader Willi syndroom). Imprinting is het gevolg van structuurveranderingen van het DNA. Methylering speelt hierin een belangrijke rol. Een afwijkende methylatie kan niet worden opgespoord met sequentie analyse. Hiertoe is aparte analyse van methylatie patronen nodig. Eén van deze methoden berust op behandeling met bisulfiet. Onder invloed van bisulfiet worden cytosines in het DNA omgezet in uracil. Dat gebeurt niet met een cytosine dat gemethyleerd is. Door eerst het DNA te behandelen met bisulfiet en vervolgens sequentie analyse uit te voeren kan onderscheid worden gemaakt tussen gemethyleerde en niet gemethyleerde delen van het DNA. Als in een bepaalde, bekende ge-imprinte regio van het genoom het methylatiepatroon op beide allelen gelijk is, of anderszins afwijkt van het normale patroon, kan de relatie hiervan met het klinisch beeld van de patiënt verder worden geanalyseerd.
-
NIPT
De Niet-Invasieve Prenatale Test (NIPT) is een bloedtest waarbij DNA in het bloed van de zwangere wordt onderzocht op chromosoomafwijkingen. Het plasma van de zwangere bevat vrij circulerend DNA (cfDNA). Deze DNA-fragmenten (gemiddeld zo’n 150 baseparen lang) zijn voor zo’n 90% afkomstig van de zwangere, ongeveer 10% is afkomstig van de placenta (dus niet direct van de foetus zelf).
Bij NIPT maakt men gebruik van zogeheten “Massively parallel sequencing”. Dit is een NGS techniek waarbij van vele honderden tot miljoenen DNA fragmenten tegelijk de basepaar volgorde wordt bepaald en de hoeveelheid kopieën wordt gekwantificeerd. Door de hoeveelheid cfDNA fragmenten per chromosoom onderling te vergelijken en te vergelijken met het aantal fragmenten van een referentie sample kan worden vastgesteld of er van bepaalde (delen van) een chromosoom meer of juist minder fragmenten zijn afgelezen. Bij een trisomie verwacht je meer fragmenten afkomstig van het betreffende chromosoom, bij een monosomie minder.
De NIPT is bedoeld als screening op trisomie 13, 18 en 21. Bij de analyse worden in principe alle chromosomen (behalve de geslachtschromosomen) geanalyseerd.Door deze genoombrede aanpak kunnen ook andere chromosoomafwijkingen worden opgespoord (zogeheten nevenbevindingen). Een zwangere kan aangeven of zij geïnformeerd wil worden over nevenbevindingen. Wil zij dit niet, dan wordt de analyse zo verricht dat alleen de uitslag voor chromosoom 13, 18 en 21 te zien is voor het laboratorium. In Nederland wordt met de NIPT niet naar de geslachtschromosomen gekeken. Het geslacht, maar dus ook eventuele afwijkingen aan de geslachtschromosomen (bijv. Turnersyndroom) zijn dus niet bekend. Meestal is het placentaire DNA identiek aan het foetale DNA. Er kan echter sprake zijn van een placentamozaïek; de chromosoomafwijking is dan in een deel van de placenta aanwezig, maar niet in de foetus. Dit kan leiden tot een fout-positieve uitslag. Een afwijking bij de NIPT moet daarom altijd met invasieve diagnostiek worden bevestigd. De test is veilig en betrouwbaar voor screening op trisomie 21, 18 en 13 in zowel hoog- als laag-risico zwangerschappen. De positief voorspellende waarde van de test is afhankelijk van het de a priori kans, de negatief voorspellende waarde is nagenoeg 100%.
Omdat een deel van het cfDNA afkomstig is van de zwangere kan met de NIPT onbedoeld ook een chromosoomafwijking bij de zwangere worden vastgesteld.
NIPT onderzoek kan worden uitgevoerd vanaf 11 weken zwangerschap.
Alle kenmerken van NIPT op een rij:
- Onderzoek op chromosomaal niveau, resolutie >20Mb
- Genoombreed
- Indicatie: screening op trisomie 21, 18 en 13 bij de foetus, vroeg in de zwangerschap. Ook partiële deleties en duplicaties kunnen worden gevonden
- Dit onderzoek toont niet aan:
- Afwijkingen aan de geslachtschromosomen
- Afwijkingen aan chromosomen anders dan 13, 18 en 21 als door de zwangere gekozen is om niet geïnformeerd te willen worden over nevenbevindingen
- Genmutaties
- Afwijkingen door repeat verlenging (vb Fragiele X syndroom)
- Afwijkingen door imprintings-/methyleringsdefecten
- Afwijkingen in mitochondrieel DNA
- Omdat het (niet-maternale) DNA dat wordt geanalyseerd niet direct afkomstig is van de foetus, maar van de placenta, moet een afwijkende bevinding altijd worden bevestigd met invasief onderzoek.
Meer informatie over de NIPT op de website van het RIVM.
-
Diagnostiek op tumorweefsel
Zie de pagina Leidraad voor verwijzing na DNA-onderzoek in (tumor)weefsel.